Хроматографический анализ эфирных и жирных масел.
Хроматографический анализ эфирных масел.
Является наиболее перспективным и наиболее информативным при исследовании эфирных масел. Газо-жидкостная хроматография (ГЖХ) – это эффективный способ разделения и установления количественных отношений в составе эфирных масел.
Для проведения хроматографии с помощью микрошприца в узкую кварцевую капиллярную трубку (внутренний диаметр 0.25 мм) длиной 30 метров испаряют (при температуре 2500С) небольшую пробу (0.001 мкл) эфирного масла. Под действием постоянно протекающего через эту трубку газа-носителя (обычно гелий, водород или азот) эфирное масло в виде пара движется по трубке. Одновременно температура колонки повышается от 500С до 2200С со скоростью 3-4 градуса/мин. Внутренняя поверхность трубки (которая называется колонка) покрыта тонким слоем (0.25 микрон) нейтральной жидкости полимерной природы (жидкая фаза, имеющая свое кодовое название, например, SE-30 или Carbowax 20M, отражающее хроматографические свойства фазы). Такая хроматография называется газо-жидкостной.
Компоненты эфирного масла имеют различное адсорбционное сродство к жидкой фазе из-за чего скорость продвижения вдоль колонки веществ, составляющих эфирное масло отличается друг от друга. В результате компоненты выходят из колонки в виде отдельных веществ. Обычное время анализа эфирного масла не более 30-40 мин. В современных методиках отдельное вещество может выходить в течение 3-7 секунд. Таким образом за время анализа можно обнаружить сотни веществ. Эти вещества различными методами детектируются.
Основным методом обнаружения веществ, выходящих из колонки является ионизационно-пламенное детектирование. С этой целью выходной конец колонки вставляется в детектор, представляющий собой тонкое сопло с непрерывно горящим пламенем водорода. Поступающие в пламя вещества ионизируются под действием высокой температуры и обнаруживаются по появлению этих ионов. Количество ионов пропорционально количеству вещества в пламени.
В результате проведения хроматографического анализа получают хроматограмму, то есть графическое изображение состава эфирного масла в виде пиков. Размеры пика указывают на количество вещества в пробе. Количественные соотношения (обычно виде процентного отношения) рассчитываются автоматически с помощью специальных калькуляторов или с помощью компьютера. На рисунке приведена хроматограмма эфирного масла из цветков хризантемы:
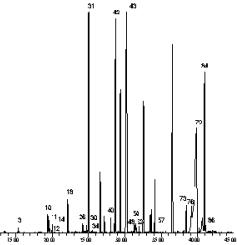
Основная проблема хроматографии – идентификация отдельных пиков, то есть расшифровка состава эфирного масла на основании данных, полученных в результате хроматографирования.
Так как время выхода различных компонентов эфирных масел отличается друг от друга, естественным способом их идентификации является использование этого времени, как постоянной для всех веществ. Использование этого метода имеет существенное ограничение, так как время выхода сильно зависит от условий хроматографирования (главным образом, давления газа и температуры) и поэтому используется очень редко в строго постоянных условиях работы хроматографической системы, с эфирными маслами, состав которых известен. Полученные результаты верны обычно только в пределах одной лаборатории и даже прибора.
Более универсальным методом использования времени как хроматографической постоянной являются индексы удерживания Ковача (Kovach's retention index). Эти индексы рассчитываются по данным времени удерживания неизвестных веществ и веществ-стандартов, для которых индексы удерживания приняты постоянными. В качестве таких стандартов выбраны нормальные алканы. Например, индекс удерживания декана (алкана с 10 атомами углерода) принят как 1000, ундекана (11 атомов углерода) - 1100 и так далее. Если теперь взять неизвестное вещество и прохроматографировать в присутствии декана и ундекана, то в случае, если это неизвестное вещество выйдет по времени между этими стандартами, то его индекс удерживания будет располагаться между 1000 и 1100. Для более точных (но упрощенных) расчетов измеряют разность времен удерживания (Δ T=T2-T1) и разность индексов удерживания двух стандартных алканов (Δ RI=RI2-RI1), а также время удерживания неизвестного вещества (T) и вычисляют его индекс удерживания (RI) по формуле:
RI=RI1+ΔRI/ΔT*(T-T1)
Индекс удерживания является характеристикой определенного вещества в условиях хроматографирования на определенной жидкой фазе. Полученные индексы удерживания имеют универсальный характер и не зависят от динамических условий хроматографирования. Если говорить более точно, условия хроматографирования (температурные градиенты в процессе анализа, природа газа-носителя) слегка изменяют значение индексов удерживания. Но эти отклонения составляют не более 0.5 единиц для неполярных фаз и не более 5-8 - для полярных фаз.
Результаты хроматографического анализа представляют в виде таблицы веществ, составляющих эфирное масло с указанием индекса удерживания для каждого вещества и процентного содержания каждого компонента.
Конечно, когда разделяемые вещества имеют близкие индексы удерживания, условия хроматографирования могут привести к наложению пиков, что хорошо заметно при работе на высокоразрешающих капиллярных колонках. Тогда возникает необходимость коррекции условий анализа. Например, на неполярной фазе SE-30 при применении газа-носителя азота происходит неполное разделение пиков пара-цимена, 1,8-цинеола и цис-β-оцимена. Использование водорода дает полное разделение этих веществ. При этом применение азота на неполярной фазе удобно для разделения триплета нерол, нераль и цитронеллол, тогда как применение водорода приводит к почти полному наложению этих пиков.
Различные режимы программирования температуры в ходе анализа также влияет на индексы удерживания. Некоторые вещества имеют "плавающие" индексы удерживания. Это происходит в случае, если проба содержит большое количество растворителя. В зависимости от величины пробы растворитель в большей или меньше степени как бы "притягивает" некоторые вещества, изменяя их индекс удерживания. Например, этилацетат в присутствии большого количества этилового спирта уменьшает свой индекс удерживания (до 10 единиц) и может совмещаться с бутанол-2 (при анализе компонентов коньячного спирта).
Обычная хроматография имеет ограниченные возможности идентификации. Появление масс-спектрометрического детектора сделало возможным проводить идентификацию компонентов эфирного масла автоматически, в ходе выполнения аналитического цикла. Такие хроматографы сочетающие обычный хроматограф (GC) и масс-спектрометрический детектор (MS) получили название хромато-масс-спектрометра (ХМС, GCMS – gas-chromatography-mass-spectrometry). Обычная стоимость хроматографа 10,000-50,000$, стоимость хромато-масс-спектрометрического детектора 150,000-250,000$. При идентификации компонентов методом MS выходящие из колонки вещества не попадают в горящее пламя водорода, а направляются в поток электронов, имеющих стандартную энергию (70 эв). Эти электроны разбивают молекулу вещества на заряженные осколки, состав, заряд и количество которых в стандартных условиях для каждого вещества является постоянным. Эта информация хранится в виде баз данных в компьютерах, обслуживающих работу GCMS. Размер такой библиотеки масс-спектров может составлять более 220000 веществ.
В процессе хроматографического разделения по мере выхода веществ из колонки происходит непрерывная их бомбардировка электронами, получение масс-спектра, сравнение его с базой данных и выдача результатов. Обычно результаты GCMS представляют собой ряд предложений по идентификации в виде альтернативных формул веществ. Задача исследователя – отобрать из них наиболее приемлемый вариант. При этом пользуются законами взаимосвязи компонентов в эфирных маслах. Например, если в эфирном масле идентифицирован линалилацетат и не найден линалоол, то это не является правильным выбором (или надо искать линалоол). Аналогично, если идентифицирован нерол, а отсутствует гераниол (эти два вещества всегда присутствуют вместе), то такая идентификация сомнительна (или надо искать гераниол). Часто тимол сопровождает карвакрол, пара-цимен и гамма-терпинен, пинокамфон - изопинокамфон и т. д. Взаимосвязи компонентов в эфирных маслах обусловлены биогенетическими связями и существующими в растениях достаточно однообразными биохимическими процессами, которые связывают все компоненты эфирных масел в единый биологический цикл. Знание таких процессов позволяет избежать ошибок при идентификации.
Частая ошибка идентификации - ложные пики, образующие как артефакты при выделении эфирных масел (остатки от предыдущей пробы, примеси в растворителях, продукты разложения жидкой фазы в колонках). Такие "находки" следует ждать всегда. По крайней мере надо хорошо представлять возможный химический состав хроматографируемого объекта и упредить появление возможных "фантомов".
Кроме того каждый компонент, идентифицированный с помощью хромато-масс-спектрометра должен быть сверен со своим индексом удерживания и лишь на основании такой всесторонней оценки делается вывод об окончательной идентификации.
Иногда для получения вспомогательной информации (для уточнения идентификации) проводят хромато-ольфактометрию профиля компонентов эфирных масел. Для этого в процессе хроматографического анализа выходной конец хроматографической колонки обнюхивают. Выходящие последовательно компоненты эфирного масла оцениваются экспертами качественно. Наиболее интересные вещества идентифицируются для возможности дальнейшего их синтеза и использования для создания парфюмерных композиций. Этим методом, удается четко и наглядно представить себе вклад отдельных составляющих эфирного масла в его аромат. Так было установлено, что в состав эфирного масла розы входят на уровне миллионных долей органические сульфиды, гнилостный запах которых иногда удается почувствовать в некоторых образцах эфирного масла розы. Не следует думать, что "плохой" запах - это плохо. Среди компонентов эфирных масел многих цветочных растений (жасмин, например) содержатся производные индола, имеющие в чистом виде запах человеческих экскрементов. Но при содержании на уровне 0.001% они имеют запах очень похожий на запах жасмина.
Большая база данных по запаху и индексам удерживания различных веществ, входящих в состав ароматических продуктов находится на сайте Mottram D.S. (университет в Рединге, Великобритания) по адресу http://www.mottram.uklinux.net/
Рассмотрим пример. Поступил образец масла жожоба (на самом деле жожоба – это не масло, а растительный воск) для проверки чистоты от других жирных масел.
Для проверки качества масла жожоба проводят разложение масла на отдельные составляющие (кислоты и спирты) и метилирование кислот методом переэтерификации 14% раствором BCl3 в безводном метаноле (методика автора).
Для этого в виалу на 2 мл наливают 1 мл метилирующего реактива (Supelco, #3-3033) и добавляют 0.01 мл (приблизительно, взятую на шток от микрошприца маленькую каплю) масла жожоба.
Нагревают реакционную смесь в виале, плотно закрытой тефлоновой крышкой, при 80 град в течение около часа (я это делаю в термостате хроматографа). После того как капля растворится (смотрят в лупу, чтобы убедиться, что в реакционной смеси нет эмульсионных включений) виалу охлаждают и реакционную смесь нейтрализуют 0.3 мл 5% раствором NaOH, контролируя нейтрализацию по универсальной бумажке.
Процесс нейтрализации имеет критическое значение для дальнейшего хроматографического процесса. Теоретически, после окончания переэтерификации смесь можно впрямую хроматографировать. Но при этом надо помнить, что в ней содержится много активной соляной кислоты, которая достаточно быстро (за один цикл анализа) может разрушить связи привитой фазы. В результате капиллярная колонка становиться «активной» и малоэффективной.
Раствор при нейтрализации становится молочно-мутным. После нейтрализации в него добавляют 0.2-0.3 мл хлороформа и слегка встряхивают. При этом метиловые эфиры жирных кислот и высшие ненасыщенные спирты (характерные для жожоба) переходят в раствор хлороформа.
Раствор метиловых эфиров жирных кислот и свободных высших спиртов в хлороформе отбирают микрошприцом из нижнего слоя и хроматографируют на колонке INNOWAX (Agilent Technologies) или на любой другой полярной колонке.
Температура самплера 270 град.
Температуру термостата программируют от 100 до 230 град (5 град/мин).
Объем образца – 0.2 мкл.
Хроматограмма представленного образца масла жожоба
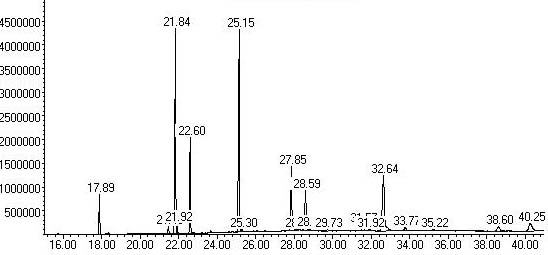
Состав смеси масел (метиловые эфиры указанных жирных кислот и свободные спирты, %%)
|
17.89
|
Пальмитиновая кислота 16:0
|
3.60
|
|
21.46
|
Стеариновая кислота 18:0
|
0.81
|
|
21.84
|
Олеиновая кислота 18:1
|
23.95
|
|
21.92
|
Элаидиновая кислота 18:1
|
0.84
|
|
22.60
|
Линолевая кислота 18:2
|
8.98
|
|
25.15
|
Эйкозеновая кислота 20:1
|
23.82
|
|
27.85
|
Эйкозеновый спирт 20:1
|
9.51
|
|
28.14
|
Докозановая кислота 22:0
|
0.35
|
|
28.59
|
Докозеновая кислота 22:1
|
6.03
|
|
32.64
|
Докозеновый спирт 22:1
|
13.61
|
|
40.25
|
Тетракозеновый спирт 24:1
|
3.34
|
|
|
Всего идентифицированных
|
94.84
|
|
|
Неидентифицированные вещества
|
5.16
|
Результаты показывают, что в смесь входят жирные кислоты, не характерные для масла жожоба (пальмитиновая, стеариновая и линолевая), а содержание олеиновой (18:1) непропорционально высокое по отношению к эйкозеновой кислоте (20:1).
Цитата:
Jojoba oil
This wax is fluid (melting point: about 6°C) and produced from seeds of the jojoba tree (Simmondsia chinensis, Euphorbiacae), now cultivated in Mexico, Arizona and California.
It is formed quite exclusively of alcohols esterified with long-chain fatty acids (more than 98%) with a total of 38 to 44 carbon atoms. The fatty acids are 18:1 (about 10%), 20:1 (about 70%) and 22:1 (15-20%), while the fatty alcohols have predominantly 20 and 22 carbon atoms and one double bond. Jojoba oil is very resistant to oxidation and is largely used in cosmetic (shampoos, anti-solar oils). Industries use sulfonated or hydrogenated oil as lubrificant.
В составе смеси мы легко находим указанные в цитате специфические компоненты масла жожоба, которые содержатся (в %% от общего состава)
|
Эйкозеновая кислота 20:1
|
23.82
|
|
Эйкозеновый спирт 20:1
|
9.51
|
|
Докозановая кислота 22:0
|
0.35
|
|
Докозеновая кислота 22:1
|
6.03
|
|
Докозеновый спирт 22:1
|
13.61
|
|
Тетракозеновый спирт 24:1
|
3.34
|
|
Всего компонентов жожоба в исследуемой смеси
|
6.66
|
Кроме того, в состав жожоба (по литературным данным) входит олеиновая кислота. По отношению к другим компонентам ее содержание (по литературным данным) составляет приблизительно седьмую часть от эйкозеновой кислоты (см. цитату), то есть около 3.4% (23.95/7). Отнимаем эту величину от общего содержания олеиновой кислоты (23.95-3.4). Получаем полный относительный состав масло жожоба и абсолютный (приведенный к 100%).
|
Олеиновая кислота 18:1
|
3.40
|
5.66
|
|
Эйкозеновая кислота 20:1
|
23.82
|
39.66
|
|
Эйкозеновый спирт 20:1
|
9.51
|
15.83
|
|
Докозановая кислота 22:0
|
0.35
|
0.58
|
|
Докозеновая кислота 22:1
|
6.03
|
10.04
|
|
Докозеновый спирт 22:1
|
13.61
|
22.66
|
|
Тетракозеновый спирт 24:1
|
3.34
|
5.56
|
|
Всего компонентов жожоба в смеси
|
60.06
|
100.00
|
Состав компонентов примеси масла выглядит следующим образом (с учетом вычтенного содержания олеиновой кислоты, принадлежащей жожоба и приведенной к 100%)
|
Пальмитиновая кислота 16:0
|
3.60
|
10.35
|
|
Стеариновая кислота 18:0
|
0.81
|
2.33
|
|
Олеиновая кислота 18:1
|
20.55
|
59.09
|
|
Элаидиновая кислота 18:1
|
0.84
|
2.42
|
|
Линолевая кислота 18:2
|
8.98
|
25.82
|
|
|
34.78
|
100.00
|
Для поиска подобия указанного состава жирных кислот другим маслам использовали небольшую базу данных автора по составу некоторых растительных масел и пакет STATISTICA6.
Данные по составу жирных кислот различных масел находятся в исходной матрице, которую при необходимости можно дополнить как известными, так и неизвестными маслами. После выполнения кластерного анализа неизвестное масло «ложиться» рядом с тем маслом, состав которого наиболее близок к нему.
Пример исходного взаиморасположения различных жирных масел известного состава жирных кислот:
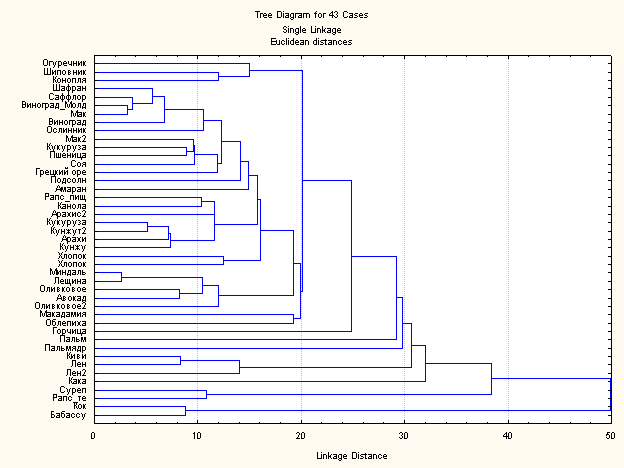
Диаграмма показывает степень сходства состава жирных кислот различных известных жирных масел.
После помещения в матрицу новой строки с неизвестным маслом (С-44) диаграмма сходства выглядит следующим образом:
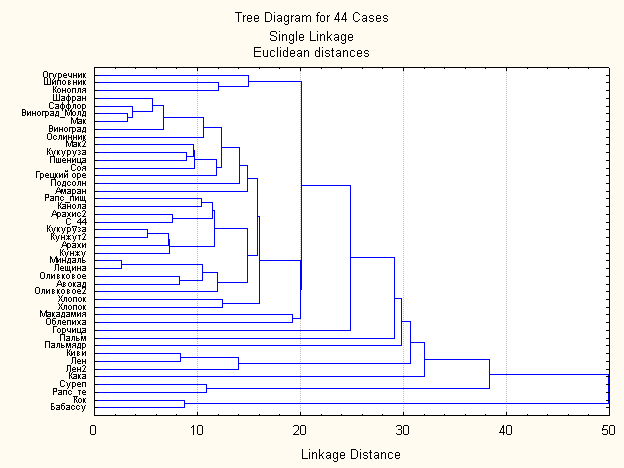
Как видно из диаграммы состав неизвестного масла (С-44) находится в кластере арахис2 и таким образом наиболее близок к нему по составу, что делает возможность сделать предположение о том, что наше неизвестное масло (которым разбавили масло жожоба) является арахисовым.
Также можно получить количественную оценку связи (которая является скорее степенью вероятности такого подобия). Для этого строится матрица расстояний (из меню «кластерный анализ» пакета STATISTICA), которая после выполнения всех вычислений выглядит следующим образом (в той части, которая касается нашего вопроса сходства):
|
C-44
|
0.0
|
|
Арахис2
|
7.6
|
|
Рапс_пищ
|
11.4
|
|
Арахис
|
14.5
|
|
Канола
|
14.8
|
|
Лещина
|
14.8
|
|
Миндаль
|
15.1
|
|
Оливковое
|
16.0
|
|
Авокадо
|
17.9
|
|
Кунжут2
|
20.7
|
|
Кукуруза
|
23.5
|
|
Оливковое2
|
25.0
|
|
Кунжут
|
28.5
|
|
Макадамия
|
31.7
|
|
Горчица
|
38.1
|
|
Подсолнечн.
|
38.2
|
|
Облепиха
|
39.1
|
|
Пальмовое
|
41.9
|
|
Хлопковое
|
42.6
|
|
Соя
|
43.8
|
|
Амарант
|
45.0
|
|
Кукуруза
|
46.0
|
|
Какао
|
46.6
|
|
Пшеница
|
48.7
|
|
Мак2
|
49.9
|
|
Огуречник
|
50.5
|
|
Хлопок
|
52.2
|
|
Пальмядровое
|
54.9
|
|
Рапс_технич
|
55.4
|
|
Грецкий орех
|
58.7
|
|
Конопля
|
60.1
|
|
Виноград
|
60.2
|
|
Шиповник
|
60.8
|
|
Суреп
|
62.1
|
|
Шафран
|
64.1
|
|
Лен2
|
65.4
|
|
Саффлор
|
65.9
|
|
Мак
|
67.2
|
|
Виноград_Молд
|
67.9
|
|
Ослинник
|
70.2
|
|
Бабассу
|
74.9
|
|
Лен
|
76.6
|
|
Кокос
|
79.4
|
|
Киви
|
81.6
|
Чем меньше значение расстояния, тем больше сходство химического состава масел. В данном случае видно, что кроме арахисового масла схожим составом обладает пищевое рапсовое масло, канола, лещина, миндаль и оливковое. Более точные результаты можно получить при обстоятельном химическом анализе и выявлении уникальных химических составляющих указанных жирных масел. Но для нашего вопроса эти исследования излишни, так как стоял вопрос о чистоте масла жожоба.
Источник: http://viness.narod.ru/jojoba.htm, http://viness.narod.ru/chrom_analys.htm
Зображення




